一种检测牦牛RNA编辑位点的方法及装置与流程
本发明属于牦牛基因功能挖掘技术领域,尤其涉及一种检测牦牛rna编辑位点的方法及装置。
背景技术:
牦牛是青藏高原独特的大型动物,高原地区人民的生产生活离不开牦牛。因为牦牛独特的高原适应能力,牦牛被称为“高原之舟”,可以适应高海拔、低氧、强紫外线等恶劣的自然环境。通过遗传图谱分析发现,牦牛的基因组与北美野牛亲缘最近,因此推测两个物种最早起源于蒙古和西伯利亚寒冷地区,在地球冰川时期,一支进入青藏高原地区,另一支通过冰冻的白令海峡进入美洲。目前诸多研究表明,牦牛的这种独特的高原适应性在主要来自遗传,也就是脱氧核糖核酸(deoxyribonucleicacid,dna)和核糖核酸(ribonucleicacid,rna)序列中某些特殊基因或者集团。这些基因和集团编码的蛋白构成了多样的生物体,然而深入的基因测序使我们了解到牦牛相对于普通肉牛或者水牛,其基因组结构98%具有相似性,而对于那些差异基因的分析又不能完全解释牦牛独特的生理结构和环境适应性。因此,目前学术界将这些和环境作用的因素归为dna到rna的变异。这种变异是由于共同的dna片段,转录成rna时,受到某些小rna的调节,导致转录的rna出现点突变或者片段突变,这些突变被称为rna编辑位点(rnaeditingsites,res)。了解这些res不仅有助于解释诸多环境和基因互作的关系,还能解释同一个个体不同组织间,功能、结构、发育上的不同,有助于帮助人们了解生物不同组织的发育过程以及功能的特异性。
目前res的探测主要集中在不同组织的转录组测序技术(rna-seq)数据分析上,多数方法利用rna序列与参考基因组比对,通过多个组织或环境下的序列差异找到res位点,这些方法从原理上解释了res的发生过程,但是这些方法具有十分高的假阳性,由于测序技术带来的测序错误,会影响rna-seq数据中许多序列测序的准确性,这就会影响反转录互补脱氧核糖核酸(complementarydna,cdna)的准确率。
因此,目前用于检测牦牛中res的方法存在准确率较低的问题。
技术实现要素:
本发明实施例的目的在于提供一种检测牦牛rna编辑位点的方法,旨在解决目前用于检测牦牛中res的方法存在准确率较低的问题。
本发明实施例是这样实现的,一种检测牦牛rna编辑位点的方法,包括以下步骤:
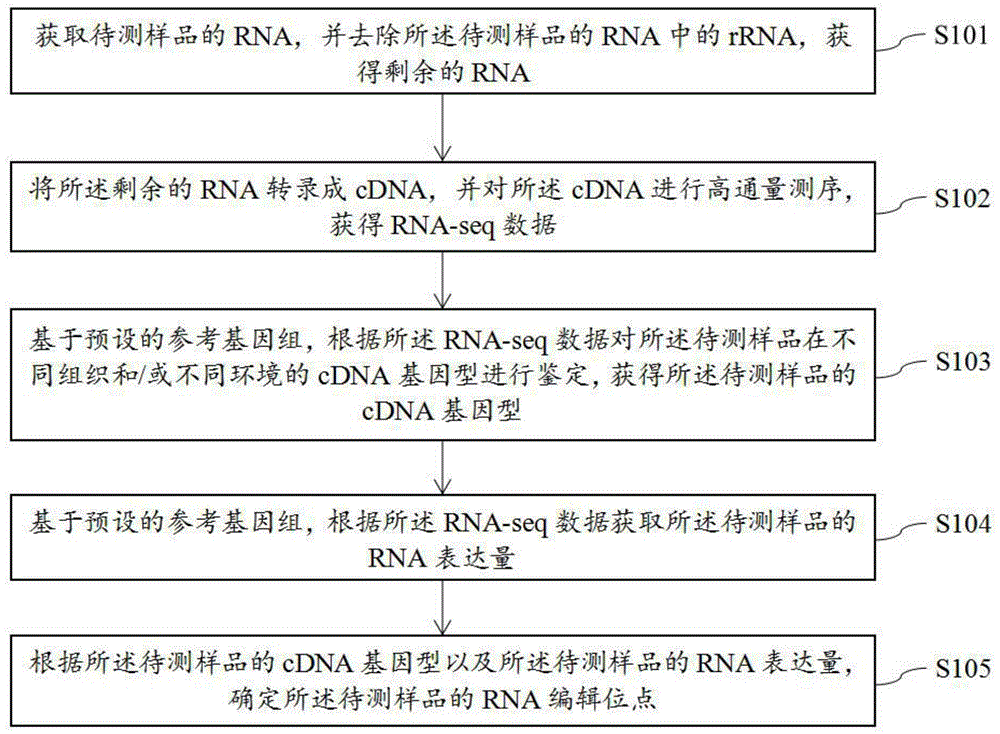
获取待测样品的rna,并去除所述待测样品的rna中的rrna,获得剩余的rna;
将所述剩余的rna转录成cdna,并对所述cdna进行高通量测序,获得rna-seq数据;
基于预设的参考基因组,根据所述rna-seq数据对所述待测样品在不同组织和/或不同环境的cdna基因型进行鉴定,获得所述待测样品的cdna基因型;
基于预设的参考基因组,根据所述rna-seq数据获取所述待测样品的rna表达量;
根据所述待测样品的cdna基因型以及所述待测样品的rna表达量,确定所述待测样品的rna编辑位点。
本发明实施例的另一目的在于提供一种检测牦牛rna编辑位点的装置,其包括:
数据获取单元,用于获取待测样品的rna-seq数据;
基因型鉴定单元,用于基于预设的参考基因组,根据所述rna-seq数据对所述待测样品在不同组织和/或不同环境的cdna基因型进行鉴定,获得所述待测样品的cdna基因型;
表达量鉴定单元,用于基于预设的参考基因组,根据所述rna-seq数据获取所述待测样品的rna表达量;
位点确定单元,用于根据所述待测样品的cdna基因型以及所述待测样品的rna表达量,确定所述待测样品的rna编辑位点。
本发明实施例提供的一种检测牦牛rna编辑位点的方法,该检测方法包括以下步骤:获取待测序样品的总rna,并根据牦牛参考基因组中rrna的数据去除待测样品总rna中的rrna;将剩余的rna反转录成cdna,随机打断cdna成片段状并进行高通量测序,获得总的粗数据;通过对粗数据的去重复、一定的质量控制和筛选,最终比对到牦牛参考基因组上,获得不同环境或不同组织上的反转录基因型数据;通过对粗数据进行质量控制和筛选,对比到牦牛参考基因组上,获得在不同环境或组织上的基因表达数据;根据待测样品的反转录基因型以及待测样品的rna表达量,获得待测样品的候选rna编辑位点;最后通过rna编辑位点的蛋白编码能力改变鉴定rna编辑位点的变异能力,最终确定有效变异的rna编辑位点。本发明综合反转录基因型和基因表达量,缩小候选rna编辑位点的范围,并利用编码蛋白能力的改变来推测有效变异,使结果更加可信,以排除假阳性位点,提高了预测rna编辑位点的准确性。本发明实施例提供的检测方法除了需要参考基因组和rna-seq数据之外,不需要额外的数据,其在方法应用上给用户带来了很大的方便。另外,本发明实施例从原理上拓展了rna编辑位点的检测方法,利用表达量对候选位点进行过滤,使结果更加可信,以排除假阳性位点,提高了预测rna编辑位点的准确性。其中,该rna编辑位点的检测方法可有效探测组织间和多环境下牦牛的rna编辑位点,从而可以帮助遗传学者研究基因与环境互作的关系。
附图说明
图1为本发明实施例提供的一种检测牦牛rna编辑位点的方法的流程图;
图2为本发明实施例提供的步骤s103的流程图;
图3为本发明实施例提供的步骤s104的流程图;
图4为本发明实施例提供的步骤s105的流程图;
图5为本发明实施例提供的另一种检测牦牛rna编辑位点的方法的流程图;
图6为本发明实施例提供的一种检测牦牛rna编辑位点的装置的结构框图;
图7为本发明实施例提供的基因型鉴定单元的结构框图;
图8为本发明实施例提供的表达量鉴定单元的结构框图;
图9为本发明实施例提供的位点确定单元的结构框图;
图10为本发明实施例提供的另一种检测牦牛rna编辑位点的装置的结构框图;
图11为三种方法预测res编码蛋白能力的文氏图。
具体实施方式
为了使本发明的目的、技术方案及优点更加清楚明白,以下结合附图及实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。
可以理解,本申请所使用的术语“第一”、“第二”等可在本文中用于描述各种元件,但除非特别说明,这些元件不受这些术语限制。这些术语仅用于将第一个元件与另一个元件区分。举例来说,在不脱离本申请的范围的情况下,可以将第一xx脚本称为第二xx脚本,且类似地,可将第二xx脚本称为第一xx脚本。
如附图1所示,图1为本发明实施例提供一种检测牦牛rna编辑位点的方法的流程图,其包括以下步骤:
步骤s101,获取待测样品的rna,并去除所述待测样品的rna中的rrna,获得剩余的rna;
步骤s102,将所述剩余的rna转录成cdna,并对所述cdna进行高通量测序,获得rna-seq数据;
步骤s103,基于预设的参考基因组,根据所述rna-seq数据对所述待测样品在不同组织和/或不同环境的cdna基因型进行鉴定,获得所述待测样品的cdna基因型;
步骤s104,基于预设的参考基因组,根据所述rna-seq数据获取所述待测样品的rna表达量;
步骤s105,根据所述待测样品的cdna基因型以及所述待测样品的rna表达量,确定所述待测样品的rna编辑位点。
具体的,在步骤s101和s102中,首先对待测样品进行消毒、收集和总rna提取,需要说明的是,对于总rna的提取办法可以采用现有技术,对此没有限制;获得总rna后,通过参考基因组的核糖体rna比对,去掉rrna,其中,牦牛的参考基因组可以采用现有的gcf_000298355.1bosgruv2.0,其可以通过在ncbi网站上下载得到;然后,通过反转录试剂将剩余rna转录成cdna,并通过物理手段随机打断成200bp以下的小片段;便可在测序平台对打断得到的小片段进行高通量测序,以获得待测样品的rna-seq数据。另外,对于rna-seq数据,还需要根据测序数据质量要求进行数据质量控制,其中具体数据质量要求如下:q20数据要求大于95%,q30数据要求大于90%,片段比对率达到95%以上,牦牛数据中gc含量(指牦牛基因数据中agct四种碱基gc所占得比例)应达到40%以上。
如附图2所示,作为本发明实施例的一个优选方案,所述基于预设的参考基因组,根据所述rna-seq数据对所述待测样品在不同组织和/或不同环境的cdna基因型进行鉴定,获得所述待测样品的cdna基因型的步骤s103,具体包括:
步骤s201,滤除所述rna-seq数据中的重复序列,获得滤除后的rna-seq数据;
步骤s202,将所述滤除后的rna-seq数据中的序列按照染色体顺序进行排列,生成滤除后的基因组;
步骤s203,将所述滤除后的基因组与所述预设的参考基因组进行比对,生成第一比对数据;
步骤s204,根据所述第一对比数据对所述待测样品在不同组织和/或不同环境的cdna基因型进行鉴定,获得所述待测样品的cdna基因型。
具体的,步骤s103的主要是为了快速鉴定同一个个体不同组织或者不同环境下cdna的基因型。其中,rna-seq数据转单核苷酸多态性(singlenucleotidepolymorphism,snp)基因型时,首先需要去掉重复序列,并且通过samtools软件按照染色体顺序排列生成滤除后的基因组,该基因组可以通过sam和bam文件保存;随后利用预设的牦牛参考基因组(gcf_000298355.1bosgruv2.0)进行基因组比对,生成第一比对数据;最后,根据第一比对数据整理所有组织或环境下cdna基因型,生成待测样品的cdna基因型,可以以vcf文件进行保存,其包含不同组织或者不同环境下的cdna基因型。
如附图3所示,作为本发明实施例的另一个优选方案,所述基于预设的参考基因组,根据所述rna-seq数据获取所述待测样品的rna表达量的步骤s104,具体包括:
步骤s301,将所述rna-seq数据中的基因与所述预设的参考基因组进行比对,生成第二比对数据;
步骤s302,根据所述第二比对数据,计算所述待测样品与所述预设的参考基因组的相对表达量;
步骤s303,根据所述待测样品与所述预设的参考基因组的相对表达量,获得所述待测样品的rna表达量。
具体的,步骤s104主要用来组装转录本,并计算各个基因的表达量。首先直接将步骤s102获取的rna-seq数据进行参考基因组比对,参考基因组同样可以采用现有牦牛参考基因组(gcf_000298355.1bosgruv2.0),比对后的结果需要拼装转录本并计算相关表达量,这里可以使用tpm(transcriptspermillion)进行定义相对表达量,tpm的计算方法为现有技术,在这边就不作详细赘述,该方法可以在保证多样本之间总表达量一致的同时,将基因直接的表达量描述清楚。
如附图4所示,作为本发明实施例的另一个优选方案,所述根据所述待测样品的cdna基因型以及所述待测样品的rna表达量,确定所述待测样品的rna编辑位点的步骤s105,具体包括:
步骤s401,根据所述待测样品的cdna基因型,滤除所述待测样品中在不同组织和/或不同环境下具有相同的单核苷酸多态性和/或拷贝数变异的位点以及未知基因型的位点,以总体变异率1%作为阈值筛选候选rna编辑位点,获得第一rna编辑位点候选群;
步骤s402,基于预设的阈值,根据所述待测样品的rna表达量滤除所述待测样品中表达不明确的位点,获得第二rna编辑位点候选群;
步骤s403,根据所述第一rna编辑位点候选群以及所述第二rna编辑位点候选群,确定所述待测样品的rna编辑位点。
其中,在步骤s401中,需要对待测样品的cdna基因型数据中的单核苷酸多态性和拷贝数变异进行初步滤除,具体的,滤除方法遵循以下原则:过滤掉那些在所有组织或者环境下相同的单核苷酸多态性或者拷贝数变异以及过滤掉那些未知基因型的数据(测序数据基因型会经常出现na,即缺失的情况,这种情况可能是测序技术还不完善,或者这个位置比较难于探测。这里我们用最严格的方式去掉na数据,而非其他方法的保留一部分再进行填充);另外,根据输入个体数量确定保留在不同组织或者环境下变异趋势一致的基因型位点,以构成第一rna编辑位点候选群(比如在三个个体上,三个组织中基因的表达趋势一致的位点需要保留下来,构成第一rna编辑位点候选群)。
此外,在步骤s402中,实际上是需要保留所有组织样本或者环境下均明确表达的基因位点,即以tpm≥1作为阈值进行过滤,滤除所述待测样品中表达不明确的位点,同时保留在不同个体之间,组织样本或者环境下相对表达量趋势一致的基因位点,即可构成第二rna编辑位点候选群。其中,第一rna编辑位点候选群和第二rna编辑位点候选群的交集,即为所确定的待测样品的rna编辑位点。
如附图5所示,作为本发明实施例的另一个优选方案,上述rna编辑位点的检测方法,还包括以下步骤:
步骤s506,基于预设的基因编译蛋白分析方法,对所述待测样品的rna编辑位点进行分析,判断所述待测样品的rna编辑位点是否会影响蛋白翻译,以确定待测样品的有效变异的rna编辑位点。
具体的,步骤s506是用于对rna编辑位点的功能进行预测和鉴定,其主要通过位点信息将整个基因的外显子从个体基因组数据中提炼出来,通过现有的基因编译蛋白分析法(该方法是基于现有的软件cnci,cpat或cpc2实现的)对这些rna编辑位点进行分析,以判断rna编辑位点是否会带来蛋白质的改变,同时给这些rna编辑位点进行评分,其评分标准主要依赖编码蛋白预测软件,这些评分用来评rna编辑位点是否影响蛋白翻译,以便于为用户下游实验提供参考。参考附图11,该图为cnci(a)、cpat(b)和cpc2(c)三种方法预测rna编辑位点编码蛋白能力的文氏图,其利用cnci、cpat和cpc2对牦牛的rna编辑位点的编码蛋白能力进行预测,其中中间三个方法交集的部分代表这三个方法均预测rna编辑位点带来了蛋白质翻译的改变。其中428个rna编辑位点是a-g变异,592个rna编辑位点是c-t变异,剩余37个rna编辑位点是缺失变异。这些位点的探测证明了rna编辑位点具有改变蛋白翻译的作用,同时也为未来rna编辑提高牦牛生产性能或解决特定遗传疾病,提供了详细的、准确的rna编辑位点参考。
如附图6所示,本发明实施例还提供了一种检测牦牛rna编辑位点的装置,其包括:
数据获取单元610,用于获取待测样品的rna-seq数据;
基因型鉴定单元620,用于基于预设的参考基因组,根据所述rna-seq数据对所述待测样品在不同组织和/或不同环境的cdna基因型进行鉴定,获得所述待测样品的cdna基因型;
表达量鉴定单元630,用于基于预设的参考基因组,根据所述rna-seq数据获取所述待测样品的rna表达量;
位点确定单元640,用于根据所述待测样品的cdna基因型以及所述待测样品的rna表达量,确定所述待测样品的rna编辑位点。
如附图7所示,作为本发明实施例的另一个优选方案,所述基因型鉴定单元620包括:
序列滤除模块721,用于滤除所述rna-seq数据中的重复序列,获得滤除后的rna-seq数据;
序列排列模块722,用于将所述滤除后的rna-seq数据中的序列按照染色体顺序进行排列,生成滤除后的基因组;
第一比对模块723,用于将所述滤除后的基因组与所述预设的参考基因组进行比对,生成第一比对数据;
基因型获取模块724,用于根据所述第一对比数据对所述待测样品在不同组织和/或不同环境的cdna基因型进行鉴定,获得所述待测样品的cdna基因型。
如附图8所示,作为本发明实施例的另一个优选方案,所述表达量鉴定单元630包括:
第二比对模块831,用于将所述rna-seq数据中的基因与所述预设的参考基因组进行比对,生成第二比对数据;
相对表达量计算模块832,用于根据所述第二比对数据,计算所述待测样品与所述预设的参考基因组的相对表达量;
表达量获取模块833,用于根据所述待测样品与所述预设的参考基因组的相对表达量,获得所述待测样品的rna表达量。
如附图9所示,作为本发明实施例的另一个优选方案,所述位点确定单元640包括:
第一位点滤除模块941,用于根据所述待测样品的cdna基因型,滤除所述待测样品中在不同组织和/或不同环境下具有相同的单核苷酸多态性和/或拷贝数变异的位点以及未知基因型的位点,以总体变异率1%作为阈值筛选候选rna编辑位点,获得第一rna编辑位点候选群;
第二位点滤除模块942,用于基于预设的阈值,根据所述待测样品的rna表达量滤除所述待测样品中表达不明确的位点,获得第二rna编辑位点候选群;
rna编辑位点确定模块943,用于根据所述第一rna编辑位点候选群以及所述第二rna编辑位点候选群,确定所述待测样品的rna编辑位点。
如附图9所示,作为本发明实施例的另一个优选方案,上述rna编辑位点的检测装置,还包括:
位点分析单元1050,用于基于预设的基因编译蛋白分析方法,对所述待测样品的rna编辑位点进行分析,判断所述待测样品的rna编辑位点是否会影响蛋白翻译,以确定待测样品的有效变异的rna编辑位点。
需要说明的是,上述各单元可以实现为一种计算机程序的形式,计算机程序可在计算机设备上运行,计算机设备的存储器中可存储组成各单元中各模块构成的计算机程序使得处理器执行上述rna编辑位点的检测方法的各个步骤。
在本发明的一个实施例中,提供了一种计算机设备,其包括存储器和处理器,所述的存储器中存储有计算机程序,所述计算机程序被所述处理器执行时,使得所述处理器执行上述rna编辑位点的检测方法的各个步骤。
在本发明的一个实施例中,提供了一种计算机可读存储介质,所述计算机可读存储介质上存储有计算机程序,所述计算机程序被处理器执行时,使得所述处理器执行上述rna编辑位点的检测方法的各个步骤。
应该理解的是,虽然本发明各实施例的流程图中的各个步骤按照箭头的指示依次显示,但是这些步骤并不是必然按照箭头指示的顺序依次执行。除非本文中有明确的说明,这些步骤的执行并没有严格的顺序限制,这些步骤可以以其它的顺序执行。而且,各实施例中的至少一部分步骤可以包括多个子步骤或者多个阶段,这些子步骤或者阶段并不必然是在同一时刻执行完成,而是可以在不同的时刻执行,这些子步骤或者阶段的执行顺序也不必然是依次进行,而是可以与其它步骤或者其它步骤的子步骤或者阶段的至少一部分轮流或者交替地执行。
本领域普通技术人员可以理解实现上述实施例方法中的全部或部分流程,是可以通过计算机程序来指令相关的硬件来完成,所述的程序可存储于一非易失性计算机可读取存储介质中,该程序在执行时,可包括如上述各方法的实施例的流程。其中,本申请所提供的各实施例中所使用的对存储器、存储、数据库或其它介质的任何引用,均可包括非易失性和/或易失性存储器。
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对本发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。
- 基因型数据缺失的填充方法、装...
- 基于布尔代数的基因处理方法、...
- 有机化合物中或与之相关的改进...
- 用于治疗抑郁的NK-1拮抗剂...
- 基于模拟的药物治疗计划的制作...
- 一次性治疗性超声波装置的制作...
- 用于介入放射疗法(近距放射疗...
- 双模式能量递送系统的制作方法
- 照明装置的制作方法
- 基于光的治疗设备和方法与流程
- 还没有人留言评论。精彩留言会获得点赞!